Local alignment


As we mentioned above, global sequence alignment algorithms align sequences over their entire lengths. You do need to think about whether that type of alignment makes sense for your sequences. For our example, where we expect each exon to be represented in the sequences and in the same order, it has worked well - however, how well do you think this approach would work with, for example, multidomain proteins that share one domain but not others, or sequences where there have been regions of duplication? A second comparison method, local alignment, searches for regions of local similarity and need not include the entire length of the sequences. Local alignment methods are very useful for scanning databases or when you do not know that the sequences are similar over their entire lengths. The wEMBOSS program water is a rigorous implementation of the Smith Waterman algorithm for local alignments [4].
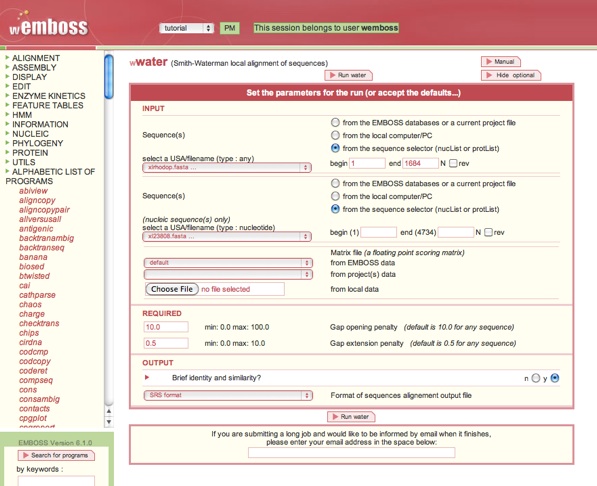
Exercise: water
Program: water
Smith-Waterman local alignment.
Input sequence: xlrhodop
Second sequence: xl23808
Gap opening penalty [10.0]:
Gap extension penalty [0.5]:
Result:
########################################
# Program: water
# Rundate: Mon 21 Apr 2008 14:12:32
# Commandline: water
# -asequence xl23808
# -sbegin1 1
# -send1 4734
# -bsequence xlrhodop
# -gapopen 10.0
# -gapextend 0.5
# -brief
# -aformat srspair
# -auto
# Align_format: srspair
# Report_file: .water.08.04.21:14.12.31/xl23808.water
########################################
#=======================================
#
# Aligned_sequences: 2
# 1: XL23808
# 2: L07770
# Matrix: EDNAFULL
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 3487
# Identity: 1683/3487 (48.3%)
# Similarity: 1683/3487 (48.3%)
# Gaps: 1804/3487 (51.7%)
# Score: 7475.0
#
#
#=======================================
XL23808 1182 gtagaacagcttcagttgggatcacaggcttctagggatcctttgggcaa 1231
||||||||||||||||||||||||||||||||||||||||||||||||||
L07770 2 gtagaacagcttcagttgggatcacaggcttctagggatcctttgggcaa 51
XL23808 1232 aaaagaaacacagaaggcattctttctatacaagaaaggactttatagag 1281
||||||||||||||||||||||||||||||||||||||||||||||||||
L07770 52 aaaagaaacacagaaggcattctttctatacaagaaaggactttatagag 101
Scroll down the entire output and again, note that five exons have been found.
In these cases we have not had to adjust the gap parameters from the defaults used in these programs. You should be aware that you might need to do so with your own sequences.
wEMBOSS contains other pairwise alignment programs - stretcher and matcher are global and local alignment programs respectively that are less rigorous than needle and water and therefore run more quickly; they may be useful for database searching. Supermatcher is designed for local alignments of very large sequences and is even less rigorous in its implementation. The documentation pages for all these programs can be found at www.emboss.org