Pairwise sequence alignment


This chapter is about sequence similarity. Let us start with a warning: there is no unique, precise, or universally applicable notion of similarity. An alignment is an arrangement of two sequences, which shows where the two sequences are similar, and where they differ. An optimal alignment, of course, is one that exhibits the most similarities, and the least differences. Broadly, there are three categories of methods for sequence comparison.
•Segment methods compare all overlapping segments of a predetermined length (e.g., 10 amino acids) from one sequence to all segments from the other. This is the approach used in dotplots.
•Optimal global alignment methods allow the best overall score for the comparison of the two sequences to be obtained, including a consideration of gaps.
•Optimal local alignment algorithms seek to identify the best local similarities between two sequences but, unlike segment methods, include explicit consideration of gaps.
Dotplots
The most intuitive representation of the comparison between two sequences uses dot-plots. One sequence is represented on each axis and significant matching regions are distributed along diagonals in the matrix.
Exercise: Making a dotplot
Program: dottup
First sequence: xl23808
Second sequence: xlrhodop
(Pay attention that the second letter after x is the letter l and not number 1 (one)
Word size [4]: 10
Graph type [png]:
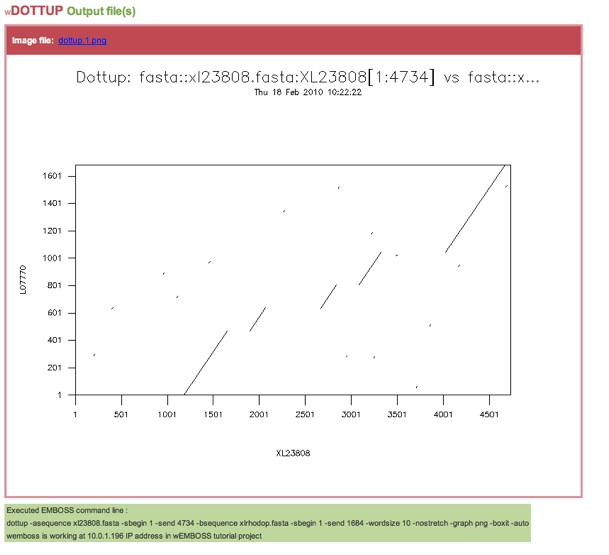
The resulting image in png format should look like this:
The diagonal lines represent areas where the two sequences align well. You can see that there are five clear diagonals. You will remember that we are aligning genomic and cDNA - these five diagonals represent the five exons of the gene! If you look at the original tembl entry for the genomic sequence using SRS, you will see that the annotated entry says that there are five exons in this gene. So our results are in agreement.
But the dotplot doesn't give us any detailed sequence information. For this, we need to use different programs. The algorithms we will be using are more rigorous than those used for searching databases; so even if you have retrieved a sequence from a database using something like BLAST, it will be well worth your while performing a careful pairwise alignment afterwards. The basic idea behind the sequence alignment programs is to align the two sequences in such a way as to produce the highest score - a scoring matrix is used to add points to the score for each match and subtract them for each mismatch. The matrices used for nucleic acid alignments tend to involve fairly simple match/mismatch scoring schemes, while the matrices commonly used for scoring protein alignments are more complex, with scores designed to reflect similarity between the different amino acids rather than simply scoring identities. Over time various mutations occur in sequences; the scoring matrices attempt to cope with mutations, but insertions and deletions require some extra parameters to allow the introduction of gaps in the alignment. There are penalties both for the creation of gaps and for the extension of existing ones; the default gap parameters given in alignment programs have been found to be empirically correct with test sequences but you should experiment with different gap penalties.
DOTPLOTS